The therapeutic window for Angelman syndrome patients may be broader than previously reported
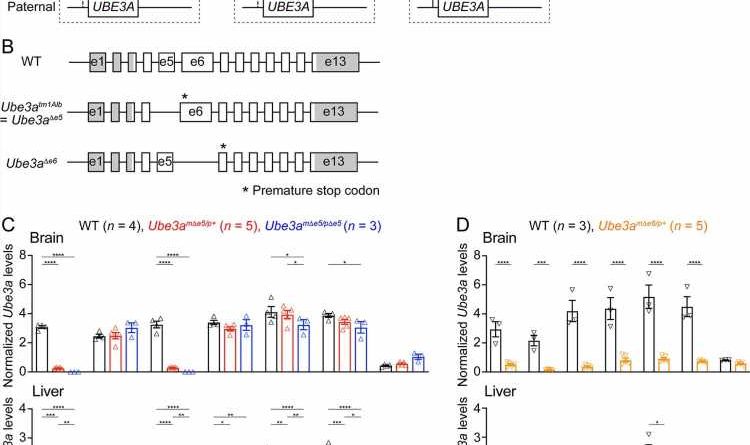
 Schematics of UBE3A imprinting and Angelman syndrome. Left, in normal neurons, UBE3A proteins are only produced from the maternal copy of UBE3A because the paternal copy is silenced by UBE3A-ATS . Middle, deficiency of the maternal UBE3A (gray) leads to the loss of UBE3A proteins in neurons and causes Angelman syndrome. Right, suppressing UBE3A-ATS expression leads to the unsilencing of the paternal UBE3A . ( B ) Genomic structures of Ube3a wild-type (WT), Δe5 (also known as tm1Alb ), and Δe6 alleles. The boxes indicate exons (e) 1–13. The white and gray regions indicate the coding and non-coding exon sequences of the longest Ube3a transcript, respectively. In the Δe5 and Δe6 alleles, exons 5 and 6 are deleted, resulting in a premature stop codon in exons 6 and 7, respectively. ( C ) Ube3a transcript levels were measured from the brains and livers of WT, Ube3a mΔe5/p+ and Ube3a mΔe5/pΔe5 mice using primer sets targeting different exons or introns as indicated in the figure. Ube3a mRNA levels were normalized by the Gapdh mRNA levels. Except the deleted exon 5, other exons in the brains of Ube3a mΔe5/p+ and Ube3a mΔe5/pΔe5 mice remain at the similar levels as WT mice. ( D ) Similar to ( C ), but for WT and Ube3a mΔe6/p+ mice. Ube3a transcript is greatly reduced in the Ube3a mΔe6/p+ mouse brains. The numbers of tested mice are indicated in the figure. Each symbol represents one mouse. Bar graphs are mean ± standard error of the mean (SEM). Two-way analysis of variance (ANOVA) with Tukey ( C ) or Šídák ( D ) multiple comparison test for all pairs of groups, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Credit: eLife (2023). DOI: 10.7554/eLife.81892")
Angelman syndrome is a rare neurodevelopmental disorder characterized by changes in brain structure, severe intellectual disability, impairments in speech, motor function, epilepsy, sleep, and unique behaviors that affects 1 in 10,000 to 24,000 people and for which no cure or treatment is currently available.
Researchers at the Jan and Dan Duncan Neurological Research Institute (Duncan NRI) at Texas Children’s Hospital and Baylor College of Medicine have now found that a gene therapy approach can effectively reverse key symptoms of this disorder in adult and juvenile rodent models, which demonstrates that the therapeutic age window for Angelman syndrome is much broader than reported previously and offers a new ray of hope for patients and their families impacted by this devastating disorder.
Moreover, the study has revealed two clinically important biomarkers that can be used to evaluate the efficacy of this and other therapeutic approaches in future clinical trials for this disorder. The study was recently published in eLife.
Angelman syndrome is caused by inactivation by the paternal Ube3a gene copy
Angelman syndrome results when the maternally-inherited copy of the UBE3A gene is either missing or defective. People normally inherit one copy of the UBE3A gene from each parent. In most tissues, both copies of the gene are turned on (active). However, in the nerve cells of the central nervous system (i.e., the brain and spinal cord), the expression of the gene copy received from the father is turned off (silenced). This phenomenon of parent-specific activation and silencing of genes is called genomic imprinting and as a result of it, neurons of the progeny have only one active copy of this gene.
If that active, maternal copy is lost due to chromosomal rearrangements or is rendered defective due to mutations, it results in the complete loss of UBE3A protein in neurons. This affects various aspects of brain development and function because this protein is critical to maintaining the balance between the synthesis and degradation of several other proteins.
Previous studies in Angelman rodent models have provided some insights into mechanisms that underlie this genetic disorder and have identified potential therapeutic strategies. A therapeutic strategy that addresses the root cause of this genetic disorder and holds great promise is the reactivation of the silenced paternal copy of the UBE3A gene using gene therapy approaches such as antisense oligonucleotides (ASOs) or CRISPR/Cas9.
Notably, previous proof-of-concept studies showed this approach was effective in restoring a few neurological symptoms in animal models of Angelman syndrome if the therapy was administered at very young ages.
“Prior studies using the ASO strategy were able to rescue some neurological symptoms in younger animals but the approach was less effective in older animals. These experiments suggested for maximum benefit, this approach would ideally have to be administered very early, perhaps even in utero in Angelman patients,” lead author Dr. Mingshan Xue, assistant professor at Baylor College and an Duncan NRI investigator, said.
“This raised concerns about its efficacy in human clinical trials because a typical Angelman patient is between one to four years of age when they receive the diagnosis of this condition. So, we decided to test if administering ASO to older Angelman mice will improve two symptoms—abnormal electrical activity of the brain and sleep disturbances—both of which are clinically very crucial but have not been studied in the context of this therapy.”
Identifying biomarkers for later intervention using a newly-generated Angelman mouse model
In this study, Xue and his team first generated a new null mouse model of Angelman syndrome. Unlike existing models of Angelman syndrome, levels of both Ube3a mRNA and protein were reduced in this new mouse model which they hypothesized would facilitate a better and more precise evaluation of gene therapy via ASOs.
They used this new mouse model to examine if gene therapy in juvenile and adult mice models of Angelman syndrome would reverse three robust and clinically critical symptoms—hyperexcitability of brain neurons (linked to seizures), abnormal electrical activity of the brain (linked to cognitive performance), and sleep disturbances—that had never been tested before.
These symptoms are closely linked to disease progression and/or severity and if this therapy was effective, the authors predicted they could act as biomarkers to guide therapy development and future clinical monitoring of a broader range of patients.
Gene therapy improves abnormal EEG patterns and sleep disturbances in the new rodent model
They found that a single injection of ASO directly into the brain of the newly-generated Angelman disease model lead to a long-lasting reactivation of the paternal copy of Ube3a throughout the brains, and an up-regulation of Ube3a protein which lasted at least 10 weeks.
As expected, the new Ube3a animal model they generated also exhibited abnormal brain activity and sleep disturbances, both of which were reversed by ASO injection and concomitant reactivation of the paternal Ube3a gene although it was not sufficient to observe any change in neuronal hyperexcitability.
“Our study has several implications for future clinical trials using this approach and others for Angelman syndrome,” Dr. Xue, who is also an investigator at the Gordon and Mary Cain Pediatric Neurology Research Foundation Laboratories and Caroline DeLuca Scholar, said.
“First, our results suggest that Angelman syndrome patients of different age ranges may benefit from this gene therapy treatment. Second, a robust correlation between the Ube3a levels and EEG power spectrum supports the notion that this metric can serve as a quantitative biomarker in future clinical trials for this disorder. Third, since the EEG power correlates closely with symptom severity, including cognitive behaviors in patients, it is reasonable to speculate that this gene therapy approach may also improve cognitive function in Angelman syndrome patients.”
“Finally, clinicians and caregivers consistently report sleep disturbances as one of the most challenging symptoms and an important consideration for new therapy development. Our gene therapy approach demonstrates a reduction in sleep disturbance in Angelman mouse models and if this finding will hold true in human trials, we are hopeful this gene therapy approach will significantly improve the quality-of-life of Angelman syndrome patients, their caregivers, and family members.”
More information:
Dongwon Lee et al, Antisense oligonucleotide therapy rescues disturbed brain rhythms and sleep in juvenile and adult mouse models of Angelman syndrome, eLife (2023). DOI: 10.7554/eLife.81892
Journal information:
eLife
Source: Read Full Article