pamelor recommended dosage
A recent study published in Microbial Genomics analyzed the molecular evolution of the variola virus (VARV).
Genetics & Genomics eBook

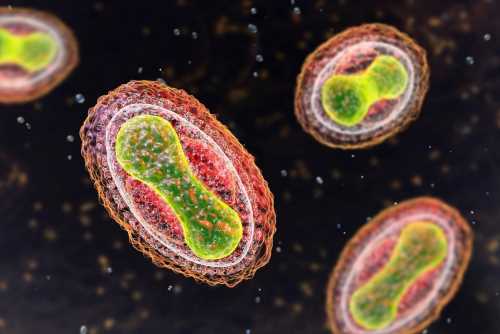
VARV, the causal pathogen of smallpox, has been a significant cause of mortality in human history. Edward Jenner pioneered vaccination for smallpox in 1796 and intensified efforts in the twentieth century to eradicate it in 1980, when only P-I and P-II lineages were circulating. VARV belongs to the Orthopoxvirus genus under Poxviridae, topiramate 200 mg weight loss which has several viruses infecting humans and mammals.
Monkeypox virus causes a similar, less severe disease than smallpox. Taterapox virus (TATV), which infects gerbils, and camelpox virus (CMLV) are most closely phylogenetically related to VARV. According to historical sources, smallpox was present in Asia and Egypt 3,500 years ago, while molecular dating suggested a more recent origin for VARV.
Genome sequencing of ancient VARV (aVARV) indicated that viral strains from the Viking age and those (historical VARV [hVARV]) from seventeenth/eighteenth-century remains were extinct. Molecular dating suggested that VARV lineages became extinct before vaccination began. It is being recognized that the time-dependent rate phenomenon (TDRP) can affect molecular dating analyses with a profound impact on events from the distant past than on recent events.
About the study
In the present study, researchers analyzed VARV genomes and re-evaluated the timeframe of its emergence by accounting for TDRP. Modern VARV (mVARV) genomes (P-I and P-II) were obtained from the National Center for Biotechnology Information (NCBI) database; those from ancient/historical remains were collected from published works. The researchers aligned whole genomes, analyzed nucleotide diversity, and computed Tajima’s D.
The population structure was analyzed taking into account biallelic parsimony-informative sites only, selected from positions with at least half the sequences having non-missing information. The team also evaluated the strength of linkage disequilibrium (LD) by testing for independent assortment by estimating the number of loci where each haplotype pair differs.
Finally, a time estimate phylogenetic reconstruction was performed. Specifically, the molecular dating of VARV was re-assessed by accounting for TDRP. To this end, the recently-developed prisoner of war (PoW) model was applied to the VARV dataset along with the whole genome sequences of CMLV and TATV.
Findings
The dataset included 48 mVARV, two hVARV, four aVARV genomes, and two specimens with controversial date estimates and analyzed 4013 parsimony-informative variants. They found a moderate level of LD, which was unlikely to be attributed to recombination since it was uncommon in the dataset. Instead, it was likely due to mutation or uneven selective pressure.
Population structure analyses revealed three sub-populations segregated as aVARV, P-I, and P-II genomes. The two controversially-dated samples clustered with P-I and P-II genomes. Conversely, hVARV genomes had the contribution of all ancestral sequences. The lowest drift from a hypothetical common ancestor was for aVARV.
The P-II lineage showed intermediate drift between P-I and aVARV. VARV lineages drifted progressively, and a minor proportion of Viking age ancestry was present until the eighteenth century. The P-I lineage experienced a more extreme bottleneck than the P-II lineage, which was confirmed by calculating nucleotide diversity and Tajima’s D.
Further, the investigators analyzed which aVARV variants were present in the hVARV sequences and found 47 variants with a high probability of aVARV ancestry in hVARV genomes. The time to the most recent common ancestor (TMRCA) for mVARV and hVARV clades and P-I and P-II clades was similar to previous estimates from other research groups.
Nevertheless, TMRCA of all VARV genomes was around 4000 years ago, and VARV split from the CMLV/TATV lineage more than 7,700 years ago. The CMLV/TATV split occurred approximately 3800 years ago. The team verified that time estimates were not influenced by the inclusion of TATV and CMLV by re-analyzing molecular dating with only VARV sequences, which revealed similar time estimates as those obtained from the extended phylogeny.
Conclusions
In summary, the time frame of the emergence of VARV and CMLV, the possibility of smallpox in ancient Egypt, and the likely presence of the ancestor of VARV, TATV, and CMLV in gerbils, are concordant with the origin of VARV in Africa or the Middle East. The study’s limitations include the limited sequences for aVARV and mVARV genomes, their biased geographic origin, and the complex passage history.
Overall, these findings provided novel insights into the origin and evolution of VARV, indicating that VARV emerged around 2,000 years before the previously estimated time, in agreement with historical records.
- Forni, D. et al. (2023) "Analysis of variola virus molecular evolution suggests an old origin of the virus consistent with historical records", Microbial Genomics, 9(1). doi: 10.1099/mgen.0.000932. https://www.microbiologyresearch.org/content/journal/mgen/10.1099/mgen.0.000932
Posted in: Medical Science News | Medical Research News | Disease/Infection News
Tags: Biotechnology, Evolution, Genome, Genomics, Monkeypox, Mortality, Mutation, Nucleotide, Pathogen, Phylogeny, Research, Smallpox, Virus

Written by
Tarun Sai Lomte
Tarun is a writer based in Hyderabad, India. He has a Master’s degree in Biotechnology from the University of Hyderabad and is enthusiastic about scientific research. He enjoys reading research papers and literature reviews and is passionate about writing.
Source: Read Full Article