pharmacy brunswick ga
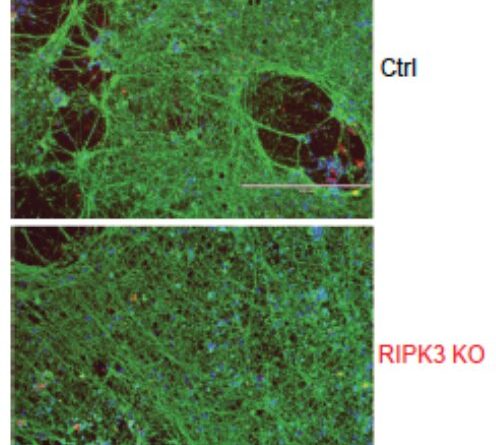
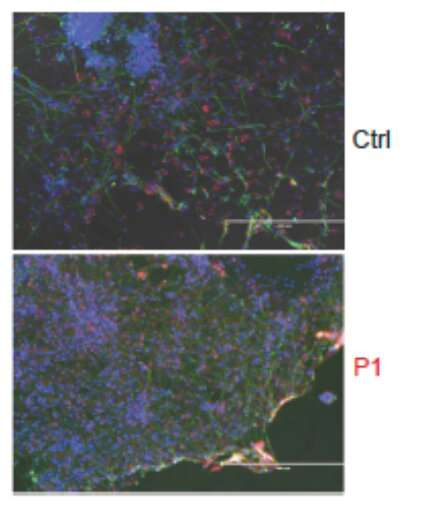
, a neuron-specific tubulin isoform (Tuj1, green) and a cortex-specific nuclear protein (TBR1, red). Credit: Science Immunology (2023). DOI: 10.1126/sciimmunol.ade2860")
Defects in a single protein involved in cell death may impair the immune system’s ability to combat a herpes viral infection in the central nervous system, scientists have found in new research that examines a pediatric infection and explains how the flawed protein can lead to a rare but severe form of encephalitis. The study is published in the journal Science Immunology.
Herpes simplex encephalitis occurs when herpes simplex virus-1 invades the brain. It is a stealthy and insidious infection, and its initial presentation is often non-specific, micronase inderal a factor that can delay diagnosis. Symptoms can include sleepiness, lethargy and fever.
As it advances, though, the infection can morph into one of the most devastating of the central nervous system despite the use of antiviral medications. In some children beset by herpes simplex encephalitis it’s not uncommon for seizures to manifest. And among affected newborns, the infection can take a rapid downhill course. Infants and older children are often exposed to the virus by close contact with someone who has a cold sore and is shedding herpes simplex-1. Other routes of infection include sharing eating utensils, towels or other items that can easily harbor the active virus.
In an unusual case study that involved a gene hunt for the DNA roots underlying susceptibility, an international team of researchers assessed the herpes simplex-1 infection of a pediatric patient who had developed herpes simplex encephalitis. Past studies had suggested that herpes simplex encephalitis in children occurs because of genetic defects in the production of—or response to—type 1 interferon, which is a key stimulator of antiviral immunity.
But Zhiyong Liu and colleagues at Rockefeller University in New York City, working with scientists at several institutions in the United States and France, found something that defied prevailing wisdom and didn’t involve genetic defects in the production, or response to, type 1 interferon.
Their first step toward discovery entailed conducting whole exome sequencing of proteins from the little girl who developed herpes simplex encephalitis. Whole exome sequencing is a genomic technique for sequencing all of the protein-coding regions of genes in a genome.
As a result of that search, the team identified two gene mutations that carry the blueprint for a key protein called RIPK3, which governs the initiation of cell death independent of type I interferon. While type 1 interferon can wage war against infiltrating viruses, a healthy RIPK3 is needed to kill off infected cells. When that doesn’t happen, the virus is free to overwhelm the central nervous system.
The finding is important because it opens a new window of understanding into what molecular mechanisms go awry in some children with herpes simplex encephalitis. The virus not only invades the brain, the infection is additionally noteworthy for its high mortality, which is estimated at roughly 20%, even when patients are treated with an antiviral. Despite earlier studies attributing susceptibility to inborn errors of immunity, which were believed to affect the production or sensing of type I interferon, the international team concluded the likely culprit is a faulty RIPK3 protein.
The U.S. Centers for Disease Control and Prevention describes herpes simplex encephalitis as the most common single cause of viral encephalitis in infants and children. Worse, whether it is treated or untreated, the infection in babies and children is inextricably linked with considerable morbidity and mortality. The collaborative group of researchers found that the flawed RIPK3 protein caused distinct cellular problems, which led to increased viral proliferation.
“The patient’s human pluripotent stem cell-derived cortical neurons displayed impaired cell death and enhanced viral growth after herpes simplex-1 infection,” Liu wrote in the Science Immunology article. The patient with herpes simplex encephalitis had compound heterozygous mutations in RIPK3, a key cytoplasmic regulator of cell death, Liu emphasized.
Yet, it’s not that defects with interferon don’t play a role at all. Apparently, inborn errors of TLR3-dependent type I interferon immunity in cortical neurons underlie forebrain herpes simplex encephalitis due to uncontrolled viral growth and subsequent cell death. What the researchers uncovered is a previously unidentified genetic link to pediatric herpes simplex encephalitis. And, at the same time, they demonstrated how cell death-dependent control is a critical component of antiviral defenses against herpes simplex-1.
“We report an otherwise healthy patient with herpes simplex encephalitis who was compound heterozygous for nonsense and frameshift RIPK3 variants,” Liu underscored, referring to his team’s findings.
The nonsense and frameshift mutations in RIPK3 constitute the two mutations discovered in the study. When RIPK3 is healthy, it is involved in initiating cell death independent of type I interferon. In their research the collaborative team found that the two mutations caused low expression of RIPK3, preventing the immune system from inducing cell death—killing infected cells—via apoptosis or necroptosis. Additionally, in the laboratory, in vitro testing revealed that fibroblasts and neurons derived from the patient could not initiate cell death, a discovery that confirmed the team’s findings.
Surprisingly, the RIPK3 deficiency did not seem to make the patient more vulnerable to other types of viral infection. “Inherited RIPK3 deficiency therefore confers a predisposition to herpes simplex encephalitis by impairing the cell death–dependent control of herpes simplex virus-1 in cortical neurons,” Liu concluded.
More information:
Zhiyong Liu et al, Encephalitis and poor neuronal death–mediated control of herpes simplex virus in human inherited RIPK3 deficiency, Science Immunology (2023). DOI: 10.1126/sciimmunol.ade2860
Journal information:
Science Immunology
Source: Read Full Article