Study reveals commercial tool used for epigenome studies is not actually appropriate for population epigenetics
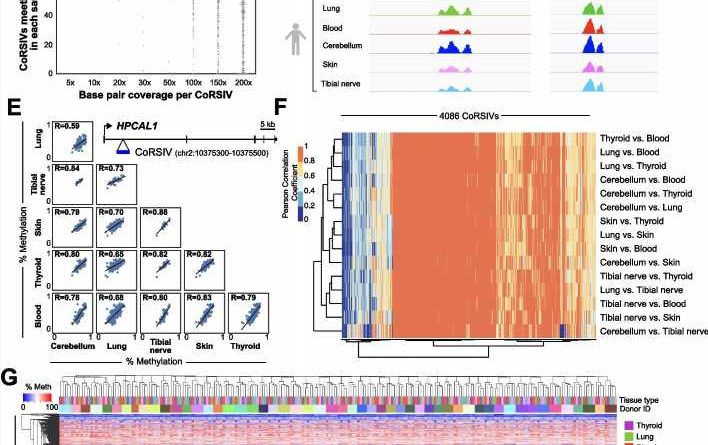
 from each of 188 GTEx donors. B CoRSIV capture process using Agilent reagents. C Percentage of CoRSIVs for which target-capture bisulfite sequencing achieved various read depths; each point represents one of 807 samples. D Plots of read depth at two target regions illustrate specificity of targeting across all six tissues. The Y-axis scales are same for each region and indicated for thyroid. E Scatter plots between all possible tissue pairs illustrate high inter-tissue correlations at a CoRSIV within HPCAL1. F Heat map of inter-tissue correlations across 4086 CoRSIVs shows generally high correlation coefficients between all possible tissue pairs. G For the 232 tissue samples from 53 donors with data on at least 4 tissues (excluding cerebellum), unsupervised hierarchical clustering of methylation data at 2349 fully informative CoRSIVs groups perfectly by donor. Credit: Genome Biology (2023). DOI: 10.1186/s13059-022-02827-3")
Twenty years ago, after the human genome was first sequenced, geneticists began conducting large genome-wide association studies to identify genomic regions linked to human disease. In addition to DNA sequence, another stable level of molecular information—epigenetic modifications established during development—can affect one’s risk of disease. For more than a decade, scientists have studied these epigenetic modifications to test associations with disease. Today more than 1,000 such epigenome-wide association studies have been published.
Now, in a study published in the journal Genome Biology, a team led by researchers at Baylor College of Medicine reveals that the commercial tool that has been the workhorse for these studies is actually not appropriate for population epigenetics.
“Many people know that each person has a unique DNA sequence or genome. Less well known is that every cell in the body likewise has a unique level of molecular individuality called its epigenome,” said co-corresponding author Dr. Robert A. Waterland, professor of pediatrics—nutrition at Baylor’s USDA/ARS Children’s Nutrition Research Center.
The epigenome—meaning “above” the genome—is a system of molecular markings on DNA that tells different cells in the body which genes to turn on or off in that cell type. “Epigenetic differences between people can affect their risk of disease,” said Waterland, a member of the Dan L Duncan Comprehensive Cancer Center at Baylor.
To look for such differences, epigeneticists study DNA methylation, which occurs at specific locations called CpG sites. The standard tool for population studies of DNA methylation is a commercial array that assays hundreds of thousands of CpG sites distributed throughout the genome.
For the last 15 years, the Waterland lab and colleagues have focused on a different set of CpG sites: those at which DNA methylation differs substantially among people but is consistent across the different tissues of each person. They reasoned that these sites would be most useful for population studies, because DNA from a blood sample can be used to investigate epigenetic causes of disease in internal organs like the brain or heart.
“Three years ago we reported nearly 10,000 such regions in the human genome (named CoRSIVs for correlated regions of systemic interindividual variation) and proposed that studying them could be a novel way to uncover epigenetic causes of disease,” Waterland said.
As a step toward this, the current study investigated how DNA methylation at CoRSIVs is affected by genetics. Correlations between a genetic variant and methylation at a specific CpG site are called methylation quantitative trait loci (mQTL). More than 200 studies of human mQTL have been reported, nearly all using the commercial methylation arrays.
The team developed an approach to target CoRSIVs and studied their methylation in DNA samples from multiple tissues of nearly 200 individuals. When they compared their results with those of the largest previous study, “what we found was somewhat of a shock,” said first author Dr. Chathura J. Gunasekara, a data analyst in the Waterland lab. “Compared to the most powerful previous study including 33,000 people, our much smaller study focused on CoRSIVs discovered 72-times more mQTL.”
Looking to explain this surprising finding, the team discovered that around 95% of the CpG sites on the commercial methylation arrays do not show appreciable methylation differences among people. Interindividual variation, which scientists call variance, is the foundation for statistical associations. With no population variance, there is no possibility of detecting mQTL.
This finding should also shock the field of epigenetic epidemiology. “Population variance is essential not only for mQTL detection, but also for detecting associations between DNA methylation and risk of disease,” said co-corresponding author Dr. Cristian Coarfa, associate professor of molecular and cellular biology and in the Dan L Duncan Comprehensive Cancer Center and the Center for Precision Environmental Health at Baylor. “Compared to what the field has been doing, we anticipate that focusing on CoRSIVs will make epigenome-wide association studies about 70 times more powerful.”
Indeed, CoRSIVs have already been associated with diverse health outcomes including thyroid function, cognition, cleft palate, schizophrenia, childhood obesity and autism spectrum disorder.
“It’s as if there’s been this massive and very expensive fishing expedition for the last 10 years, but everyone’s been fishing in the wrong place,” Waterland said. “We hope that the new tool we’ve developed will accelerate progress in understanding epigenetic causality of disease.”
More information:
Chathura J. Gunasekara et al, Systemic interindividual epigenetic variation in humans is associated with transposable elements and under strong genetic control, Genome Biology (2023). DOI: 10.1186/s13059-022-02827-3
Journal information:
Genome Biology
Source: Read Full Article