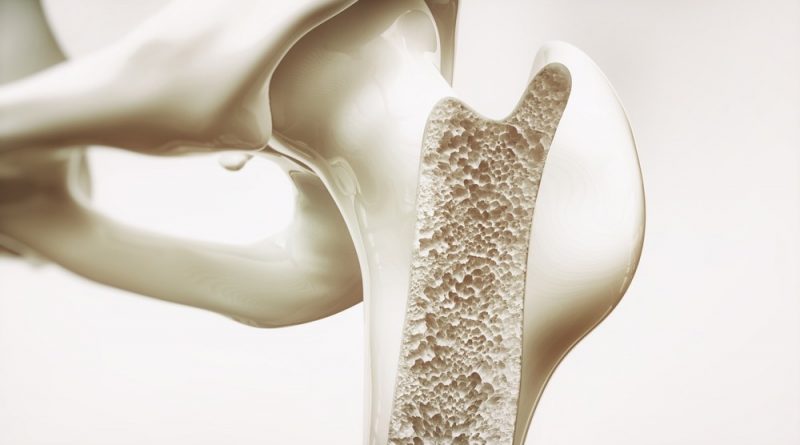
Understanding Osteopetrosis: Causes, Symptoms, Diagnosis, and Treatment
Autosomal recessive osteopetrosis (ARO)
Autosomal dominant osteopetrosis (ADO)
X-linked
Diagnosis and treatment
References
Further reading
Osteopetrosis is a rare bone disorder that occurs due to defects in osteoclast formation and function. It constitutes a group of skeletal conditions ranging from very mild to severe disease phenotypes. A rare and heterogeneous condition, osteopetrosis can provide significant difficulties for clinicians. The treatment and management of patients with osteopetrosis depend entirely on the signs and symptoms.
Image Credit: Crevis/Shutterstock.com
The German radiologist Albers-Schonberg published the first description of osteopetrosis in 1903. He described it as a heritable condition with increased bone density and a characteristic appearance of "bone inside the bone." Based on the mode of inheritance, three types of osteopetrosis can be distinguished: autosomal recessive (ARO), autosomal dominant (ADO), and X-linked.
Autosomal recessive osteopetrosis (ARO)
A severe variation of osteopetrosis, autosomal recessive osteopetrosis (ARO), first manifests itself in early infancy. Also known as malignant infantile osteopetrosis (MIOP), it might be fatal if not treated. ARO can be caused due to mutations in any of the five genes, namely, TCIRG1, CLCN7, OSTM1, SNX10, and PLEKHM1. Loss-of-function mutations in two further genes, TNFSF11 and TNFRSF11A, are also noted.
Affected people are at significant risk of suffering a bone fracture from even seemingly slight bumps and falls. In the general population, the frequency of ARO is 1 in 250 000, but it is much more common in certain geographical areas, such as the Middle East, Costa Rica, the Chuvash Republic of Russia, and the Province of Västerbotten in northern Sweden.
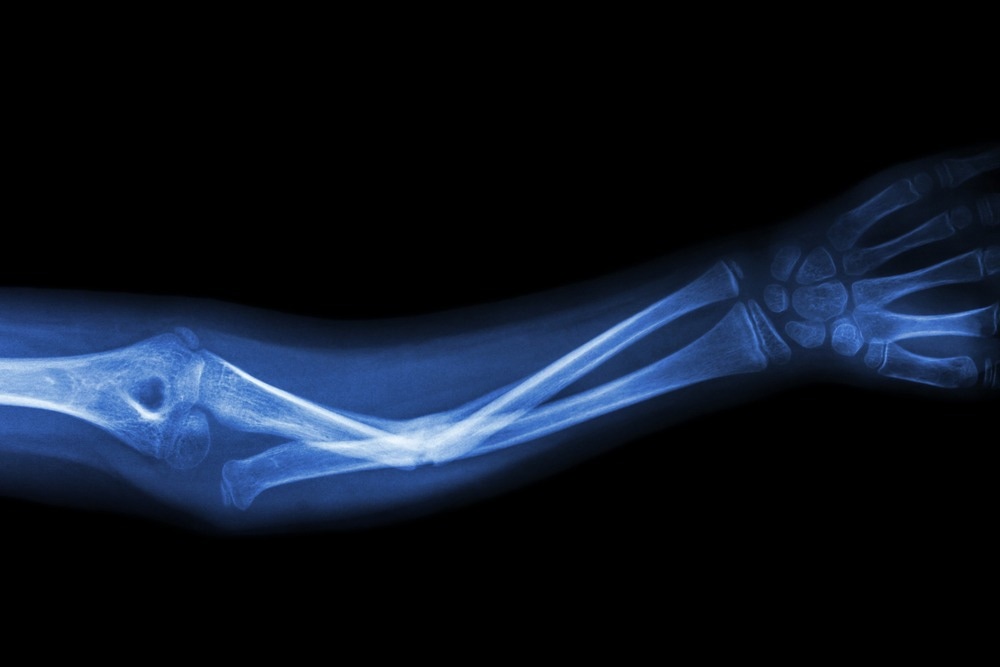
Image Credit: Puwadol Jaturawutthichai/Shutterstock.com
The primary pathological characteristics are linked to abnormal bone resorption and are present at birth or become apparent throughout the neonatal period. MIOP typically manifests as a failure to thrive, aberrant eyesight, hepatosplenomegaly, and skeletal alterations.
The cranial nerves may become encroached upon by the quickly expanding skeletal bones, leading to progressive, permanent neurologic impairment, including blindness and deafness. Visual impairment and peculiar eye motions, often known as exophthalmos, are frequently the first presenting symptoms. Delay in tooth emergence can lead to serious dental cavities.
Autosomal dominant osteopetrosis (ADO)
Albers-Schönberg disease, commonly known as autosomal dominant osteopetrosis (ADO), is often the least severe form. The overall prevalence of the disease is underestimated since so many people with ADO are asymptomatic. It is the most widespread type and can be further divided into type I and type II.
The main characteristics of the disorder include multiple bone fractures, scoliosis, or other spinal anomalies in affected persons who may have signs and symptoms. Additionally, osteomyelitis and hip arthritis can be detected. These symptoms typically surface during late childhood or adolescence. ADO occurs 1 in 20,000 births and is typically benign because the life expectancy is usually normal. ADO tends to skip a generation, and the estimated genetic penetrance is around 75%.
Clinically, ADO type 1 is typically very mildly manifested. ADO type 1 can be caused by a fault in enhanced bone production rather than a flaw in osteoclast activity, in contrast to other types of osteopetrosis. As a result, unlike other patients with osteopetrosis, these patients do not have an elevated fracture risk. ADO type 1 occurs due to mutations in the LRP5 gene. Osteosclerosis is frequently concentrated in the cranial vault in this form, and cranial nerve compression neuropathies are frequent.
ADO type 2 is the most frequent kind of osteopetrosis treated by orthopedic surgeons with a very variable history. Most individuals with this form of the disease have fairly normal life spans, general health, and physical appearances. It is caused due to genetic defects associated with the CLCN7 gene.
The diagnosis of this condition is frequently made following the clinical evaluation of a fracture that was radiographically proven to be pathologic or early-onset osteoarthritis. About 4 out of 5 of the individuals with ADO type 2 have fractures. Hip osteoarthritis, facial nerve palsy, and mandibular osteomyelitis are additional symptoms. However, 20–40% of the individuals continue to have no symptoms.
X-linked
Rarely, osteopetrosis can be inherited in an X-linked manner. The X-linked type of the condition is distinguished by lymphedema and anhydrotic ectodermal dysplasia, in addition to excessively dense bones. Additionally, immunodeficiency is observed making those who are affected experience severe recurring infections. IKBKG gene mutations cause the OL-EDA-ID subtype of X-linked osteopetrosis.
Diagnosis and treatment
Typically, the distinctive findings in radiographic imaging are used to establish a diagnosis. Physical features and molecular genetic testing can also aid in diagnosis.

Image Credit: DC Studio/Shutterstock.com
With no known cure, treatment is primarily supportive, with interprofessional care and surveillance as cornerstones. The treatment for osteopetrosis-related fractures and arthritis is provided by a skilled orthopedic surgeon. These individuals require routine dental examinations to avoid the problems of osteomyelitis, cysts, and abscesses.
The only known treatment for ARO is hematopoietic stem cell transplantation (HSCT). This is possible because HSCT improves bone remodeling, reverses pancytopenia, and promotes extramedullary hematopoiesis. However, HSCT is ineffective for osteopetrosis brought on by absent osteoclasts as a result of a mutation in TNFSF11 and is only beneficial in situations with intrinsic osteoclast abnormalities.
References
- Bailey JR, Tapscott DC. Osteopetrosis. [Updated 2022 Apr 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557529/
- 2. Penna, S., Villa, A., & Capo, V. (2021). Autosomal recessive osteopetrosis: mechanisms and treatments. Disease models & mechanisms, 14(5), dmm048940. https://doi.org/10.1242/dmm.048940
- 3. Even-Or, E., & Stepensky, P. (2021). How we approach malignant infantile osteopetrosis. Pediatric blood & cancer, 68(3), e28841. https://doi.org/10.1002/pbc.28841
- 4. Chawla, A., & Kwek, E. (2019). Fractures in patients with osteopetrosis, insights from a single institution. International orthopaedics, 43(6), 1297–1302. https://doi.org/10.1007/s00264-018-4167-5
- 5. Palagano, E., Menale, C., Sobacchi, C., & Villa, A. (2018). Genetics of Osteopetrosis. Current osteoporosis reports, 16(1), 13–25. https://doi.org/10.1007/s11914-018-0415-2
- 6. Wu, C. C., Econs, M. J., DiMeglio, L. A., Insogna, K. L., Levine, M. A., Orchard, P. J., Miller, W. P., Petryk, A., Rush, E. T., Shoback, D. M., Ward, L. M., & Polgreen, L. E. (2017). Diagnosis and Management of Osteopetrosis: Consensus Guidelines from the Osteopetrosis Working Group. The Journal of clinical endocrinology and metabolism, 102(9), 3111–3123. https://doi.org/10.1210/jc.2017-01127
Further Reading
- All Fracture Content
- Naso-Orbital-Ethmoid Fractures
Last Updated: Apr 21, 2023
Source: Read Full Article